Molecules¶
A molecular or chemical graph is a structural representation of molecule in which atoms are nodes and chemical bonds define the edges of the graph.
[1]:
import rdkit.Chem as Chem
Chem.MolFromSmiles("CN1C=NC2=C1C(=O)N(C(=O)N2C)C")
[1]:
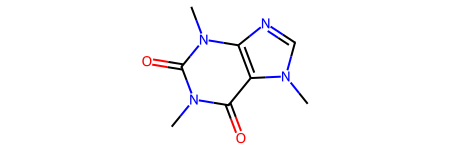
A chemical structure can be depicted in 2D with chemiformatics software such as `RDKit <https://www.rdkit.org/>`__ or `OpenBabel <https://www.rdkit.org/>`__. Both have similar class definitions of molecules, atoms and bonds classes and provide a large scope of methods to work with molecular structures.
In kgcnn.mol.base a general interface MolGraphInterface is defined that should unify access to the chemical graph used for deep learning models. Both RDKit and OpenBabel can be used as backend (Note that OpenBabel must be installed manually since no complete pypi installation is yet available).
[2]:
from kgcnn.molecule.graph_rdkit import MolecularGraphRDKit
from kgcnn.molecule.graph_babel import MolecularGraphOpenBabel
Main operations to generate a molecular graph from e.g. smiles or mol-tables as well as generating and optimizting conformation or compute charges are methods of the interface and can be used with both RDKit or OpenBabel backend. Specific kwargs for those methods, however, are handed to the specific functions of the backend and therefore require knowledge about the paramters of the backend.
[3]:
mg = MolecularGraphRDKit(make_directed=False)
mg.from_smiles("CN1C=NC2=C1C(=O)N(C(=O)N2C)C")
mg.add_hs()
mg.make_conformer()
mg.optimize_conformer(force_field="mmff94")
mg.compute_partial_charges(method="gasteiger")
[3]:
<kgcnn.molecule.graph_rdkit.MolecularGraphRDKit at 0x1dd21ec9fc0>
Note that the actual Mol-Class not privaate and accessible via mol .
[4]:
str(mg.mol)
[4]:
'<rdkit.Chem.rdchem.Mol object at 0x000001DD08843C30>'
Most importantly are methods to generate graph properties from the mol-object. In the example below are node and edge types or numbers, for the first 4 atoms or bonds, respectively:
[5]:
mg.edge_indices[:4], mg.node_symbol[:4], mg.node_coordinates[:4], mg.edge_number[1][:4] # Indices and bond order
[5]:
(array([[ 0, 1],
[ 0, 14],
[ 0, 15],
[ 0, 16]], dtype=int64),
['C', 'N', 'C', 'N'],
array([[ 3.26735076, 0.61278444, -0.2364124 ],
[ 2.1264268 , -0.26661905, -0.26747634],
[ 2.15131889, -1.61634095, -0.50052716],
[ 0.94130742, -2.13852225, -0.46338622]]),
[1, 1, 1, 1])
For attributed graphs a feature vector for atoms, bonds and the total graph has been found to improve GNNs. The functions of the interface node_attributes , edge_attributes and graph_attributes and take a list of strings (predifined) or functions for each attributes. Here are the list of attributes for RDKit by name:
[6]:
MolecularGraphRDKit.atom_fun_dict.keys()
[6]:
dict_keys(['NumBonds', 'AtomicNum', 'AtomMapNum', 'Idx', 'Degree', 'TotalDegree', 'Symbol', 'NumExplicitHs', 'NumImplicitHs', 'TotalNumHs', 'IsAromatic', 'Isotope', 'TotalValence', 'Mass', 'MassScaled', 'IsInRing', 'Hybridization', 'NoImplicit', 'ChiralTag', 'FormalCharge', 'ExplicitValence', 'ImplicitValence', 'NumRadicalElectrons', 'HasOwningMol', 'PDBResidueInfo', 'MonomerInfo', 'Smarts', 'CIPCode', 'CIPRank', 'ChiralityPossible', 'MolFileRLabel', 'GasteigerCharge', 'GasteigerHCharge', 'AtomFeatures', 'DescribeQuery', 'Rvdw', 'RvdwScaled', 'Rcovalent', 'RcovalentScaled'])
[7]:
MolecularGraphRDKit.bond_fun_dict.keys()
[7]:
dict_keys(['BondType', 'IsAromatic', 'IsConjugated', 'IsInRing', 'Stereo', 'Idx', 'BeginAtom', 'BeginAtomIdx', 'BondDir', 'BondTypeAsDouble', 'EndAtom', 'EndAtomIdx', 'Smarts', 'DescribeQuery'])
[8]:
MolecularGraphRDKit.mol_fun_dict.keys()
[8]:
dict_keys(['C', 'N', 'O', 'H', 'S', 'F', 'Cl', 'NumAtoms', 'AtomsIsInRing', 'AtomsIsAromatic', 'NumBonds', 'BondsIsConjugated', 'BondsIsAromatic', 'NumRotatableBonds', 'ExactMolWt', 'FpDensityMorgan3', 'FractionCSP3', 'MolLogP', 'MolMR', 'fr_Al_COO', 'fr_Ar_COO', 'fr_Al_OH', 'fr_Ar_OH', 'fr_C_O_noCOO', 'fr_NH2', 'fr_SH', 'fr_sulfide', 'fr_alkyl_halide'])
Then node attributes for the molecular graph can be generated from a list of attribute names.
[9]:
mg.node_attributes(["NumBonds", "Mass", "Symbol", "IsInRing", "GasteigerCharge"], encoder={})[:4]
[9]:
[[4, 12.011, 'C', False, '0.012958350519987684'],
[3, 14.007, 'N', True, '-0.32786346609948208'],
[3, 12.011, 'C', True, '0.097502273308341464'],
[2, 14.007, 'N', True, '-0.21837692759548705']]
In order to have all features of a number data type an encoder can be added for each named feature:
[10]:
from kgcnn.molecule.encoder import OneHotEncoder
mg.node_attributes(["NumBonds", "Mass", "Symbol", "IsInRing", "GasteigerCharge"],
encoder={"Symbol": OneHotEncoder(["C", "O", "N", "H"], dtype="str"), "GasteigerCharge": float})[:4]
[10]:
[[4, 12.011, 1, 0, 0, 0, 0, False, 0.012958350519987684],
[3, 14.007, 0, 0, 1, 0, 0, True, -0.3278634660994821],
[3, 12.011, 1, 0, 0, 0, 0, True, 0.09750227330834146],
[2, 14.007, 0, 0, 1, 0, 0, True, -0.21837692759548705]]
If a feature is not in the list of named attributes, a function can be inserted in place. Note that the function should always return a single value or a list of values. Argument will be the bond, molecule or atom class of the backend, respectively.
[11]:
def mol_feature(atom):
# Note that you can also get the mol object with e.g.
# mol = atom.GetOwningMol()
return atom.GetAtomicNum()
mg.node_attributes([mol_feature], encoder={})[:4]
[11]:
[[6], [7], [6], [7]]
The dataset MoleculeNetDataset uses the MolGraphInterface in map_molecule_callbacks , which specifies the used interface within mol_interface_class .
Training example¶
Example with ESOLDataset.
[12]:
%%capture
from kgcnn.data.datasets.ESOLDataset import ESOLDataset
data = ESOLDataset()
INFO:kgcnn.data.download:Checking and possibly downloading dataset with name ESOL
INFO:kgcnn.data.download:Dataset directory located at C:\Users\patri\.kgcnn\datasets
INFO:kgcnn.data.download:Dataset directory found. Done.
INFO:kgcnn.data.download:Dataset found. Done.
INFO:kgcnn.data.ESOL:Found SDF C:\Users\patri\.kgcnn\datasets\ESOL\delaney-processed.sdf of pre-computed structures.
INFO:kgcnn.data.ESOL:Read molecules from mol-file.
INFO:kgcnn.data.ESOL: ... process molecules 0 from 1128
INFO:kgcnn.molecule.encoder:OneHotEncoder Symbol found ['O', 'C', 'N', 'S', 'Cl', 'P', 'F', 'I', 'Br']
INFO:kgcnn.molecule.encoder:OneHotEncoder Hybridization found [rdkit.Chem.rdchem.HybridizationType.SP3, rdkit.Chem.rdchem.HybridizationType.SP, rdkit.Chem.rdchem.HybridizationType.SP2]
INFO:kgcnn.molecule.encoder:OneHotEncoder TotalDegree found [2, 4, 1, 3]
INFO:kgcnn.molecule.encoder:OneHotEncoder TotalNumHs found [1, 2, 0, 3, 4]
INFO:kgcnn.molecule.encoder:OneHotEncoder CIPCode found [None, 'S', 'R']
INFO:kgcnn.molecule.encoder:OneHotEncoder ChiralityPossible found [None, '1']
INFO:kgcnn.molecule.encoder:OneHotEncoder BondType found [rdkit.Chem.rdchem.BondType.SINGLE, rdkit.Chem.rdchem.BondType.TRIPLE, rdkit.Chem.rdchem.BondType.AROMATIC, rdkit.Chem.rdchem.BondType.DOUBLE]
INFO:kgcnn.molecule.encoder:OneHotEncoder Stereo found [rdkit.Chem.rdchem.BondStereo.STEREONONE, rdkit.Chem.rdchem.BondStereo.STEREOE, rdkit.Chem.rdchem.BondStereo.STEREOZ]
The above descriptors can be passed to Molecule datasets via the read_in_memory or set_attributes method.
[13]:
%%capture
from kgcnn.molecule.encoder import OneHotEncoder
data.read_in_memory(
nodes = [
'Symbol', 'TotalDegree', 'FormalCharge', 'NumRadicalElectrons', 'Hybridization',
'IsAromatic', 'IsInRing', 'TotalNumHs', 'CIPCode', "ChiralityPossible", "ChiralTag"
],
encoder_nodes = {
'Symbol': OneHotEncoder(
['B', 'C', 'N', 'O', 'F', 'Si', 'P', 'S', 'Cl', 'As', 'Se', 'Br', 'Te', 'I', 'At'],
dtype="str"
),
'Hybridization': OneHotEncoder([2, 3, 4, 5, 6]),
'TotalDegree': OneHotEncoder([0, 1, 2, 3, 4, 5], add_unknown=False),
'TotalNumHs': OneHotEncoder([0, 1, 2, 3, 4], add_unknown=False),
'CIPCode': OneHotEncoder(['R', 'S'], add_unknown=False, dtype='str'),
"ChiralityPossible": OneHotEncoder(["1"], add_unknown=False, dtype='str'),
},
edges = ['BondType', 'IsAromatic', 'IsConjugated', 'IsInRing', 'Stereo'],
encoder_edges = {
'BondType': OneHotEncoder([1, 2, 3, 12], add_unknown=False),
'Stereo': OneHotEncoder([0, 1, 2, 3], add_unknown=False)
},
graph=['ExactMolWt', 'NumAtoms'],
encoder_graph = {},
add_hydrogen=False,
make_directed=False,
has_conformers=True,
sanitize=True,
compute_partial_charges=None,
label_column_name="measured log solubility in mols per litre"
)
INFO:kgcnn.data.ESOL:Read molecules from mol-file.
INFO:kgcnn.data.ESOL: ... process molecules 0 from 1128
INFO:kgcnn.molecule.encoder:OneHotEncoder Symbol found ['O', 'C', 'N', 'S', 'Cl', 'P', 'F', 'I', 'Br']
INFO:kgcnn.molecule.encoder:OneHotEncoder Hybridization found [rdkit.Chem.rdchem.HybridizationType.SP3, rdkit.Chem.rdchem.HybridizationType.SP, rdkit.Chem.rdchem.HybridizationType.SP2]
INFO:kgcnn.molecule.encoder:OneHotEncoder TotalDegree found [2, 4, 1, 3]
INFO:kgcnn.molecule.encoder:OneHotEncoder TotalNumHs found [1, 2, 0, 3, 4]
INFO:kgcnn.molecule.encoder:OneHotEncoder CIPCode found [None, 'S', 'R']
INFO:kgcnn.molecule.encoder:OneHotEncoder ChiralityPossible found [None, '1']
INFO:kgcnn.molecule.encoder:OneHotEncoder BondType found [rdkit.Chem.rdchem.BondType.SINGLE, rdkit.Chem.rdchem.BondType.TRIPLE, rdkit.Chem.rdchem.BondType.AROMATIC, rdkit.Chem.rdchem.BondType.DOUBLE]
INFO:kgcnn.molecule.encoder:OneHotEncoder Stereo found [rdkit.Chem.rdchem.BondStereo.STEREONONE, rdkit.Chem.rdchem.BondStereo.STEREOE, rdkit.Chem.rdchem.BondStereo.STEREOZ]
Additional attributes and tensor properties can be generated with graph preprocessors. Note that there is also a graph preprocessor in kgcnn.molecule.preprocessor apart from MoleculeNetDataset .
[14]:
from kgcnn.graph.preprocessor import SetRange, SetEdgeIndicesReverse
data.map_list(SetRange(max_distance=5.0, in_place=True));
data.map_list(SetEdgeIndicesReverse(in_place=True));
data.map_list(method="count_nodes_and_edges");
[15]:
data[0].keys()
[15]:
dict_keys(['node_symbol', 'node_number', 'edge_indices', 'edge_number', 'graph_size', 'node_coordinates', 'graph_labels', 'node_attributes', 'edge_attributes', 'graph_attributes', 'range_indices', 'range_attributes', 'edge_indices_reverse', 'total_nodes', 'total_edges'])
[16]:
data.clean("edge_indices")
INFO:kgcnn.data.ESOL:Property 'edge_indices' is an empty list for graph '934'.
WARNING:kgcnn.data.ESOL:Found invalid graphs for properties. Removing graphs '[934]'.
[16]:
array([934])
Prepare graph labels
[17]:
import numpy as np
labels = np.array(data.obtain_property("graph_labels"))
if len(labels.shape) <= 1:
labels = np.expand_dims(labels, axis=-1)
Train test indices with e.g. KFold.
[18]:
from sklearn.model_selection import KFold
kf = KFold(n_splits=5, random_state=42, shuffle=True)
train_test_indices = [
[train_index, test_index] for train_index, test_index in kf.split(X=np.zeros((len(data), 1)), y=labels)]
[19]:
model_config= {
"class_name": "make_model",
"module_name": "kgcnn.literature.GIN",
"config": {
"name": "GIN",
"inputs": [
{"shape": [None, 41], "name": "node_attributes", "dtype": "float32"},
{"shape": [None, 2], "name": "edge_indices", "dtype": "int64"},
{"shape": (), "name": "total_nodes", "dtype": "int64"},
{"shape": (), "name": "total_edges", "dtype": "int64"}
],
"verbose": 50,
"cast_disjoint_kwargs": {"padded_disjoint": False},
"input_node_embedding": {"input_dim": 96, "output_dim": 64},
"depth": 5,
"dropout": 0.05,
"gin_mlp": {"units": [64, 64], "use_bias": True, "activation": ["relu", "linear"],
"use_normalization": True, "normalization_technique": "graph_batch",
"padded_disjoint": False},
"gin_args": {},
"last_mlp": {"use_bias": True, "units": [64, 32, 1], "activation": ["relu", "relu", "linear"]},
"output_embedding": "graph",
"output_mlp": {"activation": "linear", "units": 1}
}
}
[20]:
%%capture
import time
from kgcnn.models.utils import get_model_class
from keras.optimizers import Adam
from kgcnn.training.scheduler import LinearLearningRateScheduler
from kgcnn.literature.GIN import make_model
from kgcnn.data.transform.scaler.molecule import QMGraphLabelScaler
from kgcnn.data.transform.scaler.standard import StandardLabelScaler
from kgcnn.metrics.metrics import ScaledMeanAbsoluteError, ScaledRootMeanSquaredError
from datetime import timedelta
history_list, test_indices_list = [], []
model, hist, x_test, y_test, scaler, atoms_test = None, None, None, None, None, None
splits_done = 0
for i, (train_index, test_index) in enumerate(train_test_indices):
print("Running training on fold: %s" % i)
# Make the model for current split using model kwargs from hyperparameter.
# They are always updated on top of the models default kwargs.
model = make_model(**model_config["config"])
# First select training and test graphs from indices, then convert them into tensorflow tensor
# representation. Which property of the dataset and whether the tensor will be ragged is retrieved from the
# kwargs of the keras `Input` layers ('name' and 'ragged').
dataset_train, dataset_test = data[train_index], data[test_index]
x_train, y_train = dataset_train.tensor(model_config["config"]["inputs"]), labels[train_index]
x_test, y_test = dataset_test.tensor(model_config["config"]["inputs"]), labels[test_index]
atoms_test = dataset_test.get("node_number")
atoms_train = dataset_train.get("node_number")
scaler = StandardLabelScaler(with_std=True,with_mean=True, copy=True)
scaler.fit(y_train, atomic_number=atoms_train)
y_train = scaler.transform(y_train, atomic_number=atoms_train)
y_test = scaler.transform(y_test, atomic_number=atoms_test)
# If scaler was used we add rescaled standard metrics to compile.
scaler_scale = scaler.get_scaling()
mae_metric = ScaledMeanAbsoluteError(scaler_scale.shape, name="scaled_mean_absolute_error")
rms_metric = ScaledRootMeanSquaredError(scaler_scale.shape, name="scaled_root_mean_squared_error")
if scaler.scale_ is not None:
mae_metric.set_scale(scaler_scale)
rms_metric.set_scale(scaler_scale)
metrics = [mae_metric, rms_metric]
# Compile model with optimizer and loss
model.compile(loss="mean_absolute_error", metrics=metrics, optimizer=Adam(learning_rate=5e-04))
print(model.summary())
# Start and time training
start = time.process_time()
hist = model.fit(x_train, y_train,
validation_data=(x_test, y_test),
batch_size=32,
epochs=300,
validation_freq=10,
# Change to verbose = 2 to see progress
verbose=0,
callbacks= [
LinearLearningRateScheduler(
learning_rate_start=0.001, learning_rate_stop=1e-05, epo_min=100, epo=300)
])
stop = time.process_time()
print("Print Time for training: ", str(timedelta(seconds=stop - start)))
# Get loss from history
history_list.append(hist)
test_indices_list.append([train_index, test_index])
splits_done = splits_done + 1
[21]:
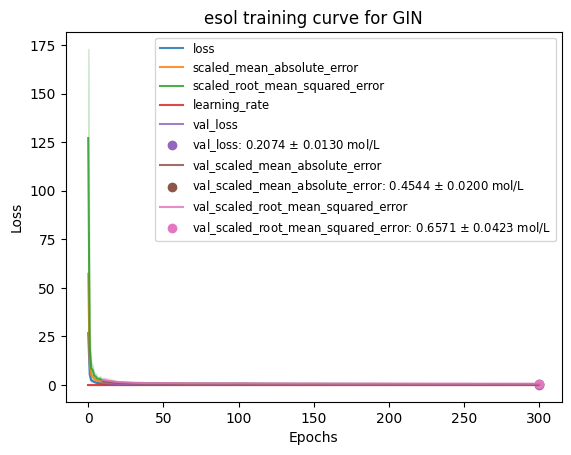
from kgcnn.utils.plots import plot_train_test_loss, plot_predict_true
plot_train_test_loss(history_list, loss_name=None, val_loss_name=None,
model_name="GIN", data_unit="mol/L", dataset_name="esol",
filepath="", file_name=f"loss.png");
[22]:
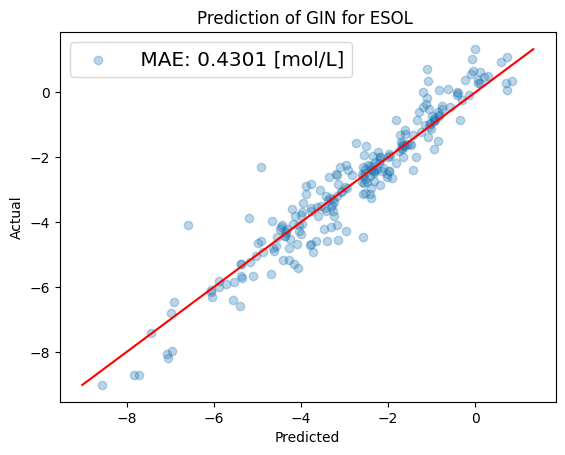
# Plot prediction for the last split.
predicted_y = model.predict(x_test)
true_y = y_test
# Predictions must be rescaled to original values.
predicted_y = scaler.inverse_transform(predicted_y)
true_y = scaler.inverse_transform(true_y)
# Plotting the prediction vs. true test targets for last split. Note for classification this is also done but
# can be ignored.
plot_predict_true(predicted_y, true_y,
filepath="", data_unit="mol/L",
model_name="GIN", dataset_name="ESOL",
file_name=f"predict.png");
8/8 ━━━━━━━━━━━━━━━━━━━━ 0s 40ms/step
NOTE: You can find this page as jupyter notebook in https://github.com/aimat-lab/gcnn_keras/tree/master/docs/source